肝豆状核变性是怎么样的病?
- 概述
-
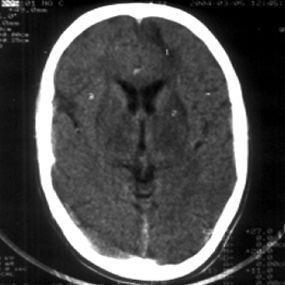
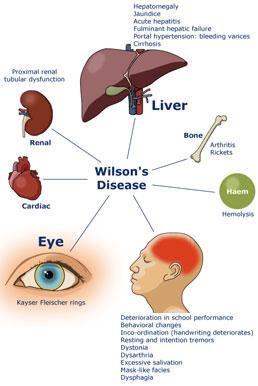
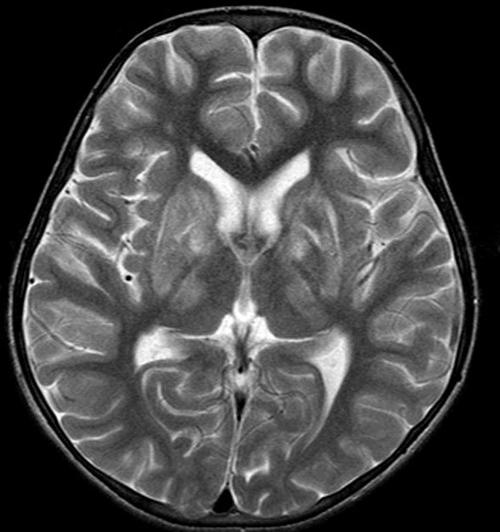
肝豆状核变性是以青少年为主的遗传性疾病,由铜代谢障碍引起。其特点为肝硬化、大脑基底节软化和变性、角膜色素环(Kayser-Fleischer环),伴有血浆铜蓝蛋白缺少和氨基酸尿症。让我们一起来分享下肝豆状核变性是怎么样的病?。
- 肝豆状核变性是怎么样的病?
-
第一:虽在婴儿期肝脏就已有铜的蓄积,但6岁前罕有肝病症状发生,而50%在15岁前发病,偶有60岁才发病者,初起症状42%为肝病表现,34%为神经系统,10%为精神症状,12%为继发于肝病的内分泌或血液系症状,1%为肾损害表现,约25%患者同时出现两个以上系统受累表现。

-
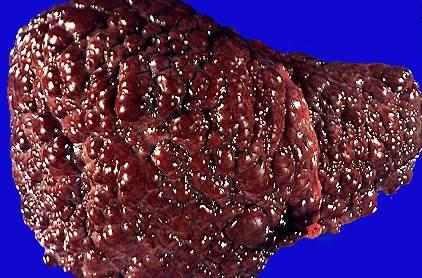
第二:以肝病为初起发病者年龄往往较小。其临床表现差异很大,可表现为急性或慢性肝炎、暴发性肝功能衰竭或肝硬化,因而,Wilson病肝脏病变所致临床表现没有特异性。在无症状期或肝硬化早期,肝功能可正常,或仅有轻微的转氨酶增高,多起病隐匿,呈现一慢性病程。开始有乏力、疲劳、厌食、黄疸、蜘蛛痣、脾肿大和脾功能亢进,最终导致门脉高压、腹水、曲张静脉出血以及肝功能衰竭。

-
第三:Wilson病患者肝脏常缩小或正常大小,有坏死后肝硬化特点,可以腹水、食管静脉曲张破裂出血为初发表现。另外临床表现、生化检查、组织学检查酷似慢性活动性肝炎者也较多见。有些患者偶尔发现K-H环或出现神经精神症状后才想到本病而得到诊断。所以,对35岁以下,HBsAg阴性的慢性肝病患者,应想到本病并做化验检查以确立诊断。

- 注意事项
-
表现多样,如肝硬化进展慢、肝外铜贮积慢,患者可多年无症状,但进展快则临床经过凶险;Ⅳ期:即络合物长期治疗后的缓解期。
- 青春无悔刚刚分享了经验 “ 早期肝纤维化治愈之路 ” 2022-08-24
- 青春无悔刚刚分享了经验 “ 肝病,早中期肝硬化必... ” 2022-08-24
- 青春无悔刚刚分享了经验 “ 大三阳患者的重生 ” 2022-08-24
- 青春无悔刚刚分享了经验 “ 【小三阳转阴成功】患... ” 2022-08-24
- 青春无悔刚刚分享了经验 “ 【大三阳成功转阴案例... ” 2022-08-23